

联系我们
地址:长沙市岳麓区枫林三路500号高盛华成2栋2单元2805
邮编:410205
联系人: 15973123761 (尹显峰)
QQ:584473091
联系人:18570397693 / 0731-83888701 (周小姐)
QQ:357013157
计算机化系统验证(Computer System Validation, CSV):建立文件来证明系统的开发符合质量工程的原则,能够提供满足用户需求的功能并且能够长期稳定工作的过程。
计算机化系统生命周期:包括由概念提出,需求理解,经由开发、放行和投入使用,直至系统退役全过程。
良好自动化生产实践指南(Good Automated Manufacturing Practice , GAMP):是由ISPE主编的实践指南。自90年代以来,不断改版的良好自动化生产实践指南被广泛使用并得到国际监管部门的公认,它是计算机化系统验证的指导方针。现行版本为第5版,即GAMP5。
GxP:基本的国际制药要求(法律或规范),包括但不限于:GMP药品生产质量管理规范、GLP良好实验室管理规范、GCP良好临床实验管理规范、GDP良好配送管理规范、GPP良好药品安全管理规范等。
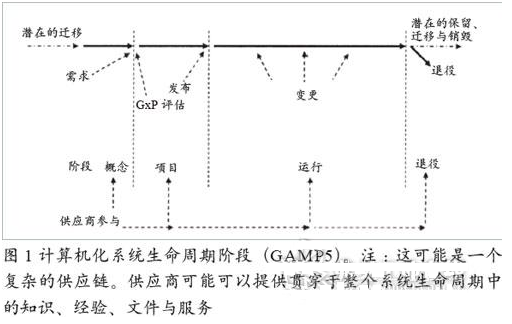
计算机化系统生命周期
计算机化系统生命周期包括从概念提出到系统退役的所有活动。由以下4个主要阶段组成:概念提出;项目实施;系统运行;系统退役。
概念提出
在概念提出阶段,公司会根据业务需求和收益来考虑是否要实现某一个或多个业务流程的自动化。通常,在这个阶段会提出初始需求并考虑可能的解决方法。通过对范围、成本和收益的初步认识,来决定是否需要进入到项目实施阶段。

项目实施
项目阶段包括以下5个方面:
计划(包括验证计划、供应商的评估和选择以及质量及项目计划);
规范(包括需求规范和设计规范);
配置和/或编程(包括源代码审核以及软硬件的集成过程);
验证(包括模块测试、集成测试和系统测试);
报告(包括验收、放行与投入使用)。
当然,项目阶段还包含风险管理、设计审查、变更和配置管理、可追溯性以及文件管理等在内的支持流程,参见图2。
系统运行
系统运行通常是最长的阶段,由既定的、及时更新的、可操作性的规程对其进行管理。保证系统处于受控(包括其安全性)、符合预期用途并且符合法规要求的状态,需要对系统进行变更和配置管理。
系统退役
当一个计算机化系统的现行功能实施不再适用,或执行一个新系统替代现有系统的功能时,该系统就从实际使用中引退。此阶段是生命周期的最后一个阶段。
计算机化系统软硬件分类
对计算机化系统进行软硬件分类也是有效的质量风险管理的一部分(软硬件类别越高,相对而言的复杂性和新颖性就越高,风险相对也就越高)。所以需要将软硬件分类同供应商评估以及GxP风险评估联系起来加以认识和理解,以上三者结合起来确定出一个适宜的验证生命周期。
值得注意的是:软硬件分类并没有特别明确的界限(尤指软件),因此并非意味着所有的软硬件均可精确地划分到某个特定的类别。
相对GAMP4、GAMP5软件分类不再单独将“类别2固件”作为一个类别,因为随着科技的发展,固件的复杂程度越来越高,可根据其嵌入软件的性质划分到任何一个类别。
硬件分为两个类别:标准硬件组件和定制硬件组件。
软件分类:GAMP5中将软件分为基础设施软件(1类)、不可配置软件(3类)、可配置软件(4类)和定制应用软件(5类)这4个类别。
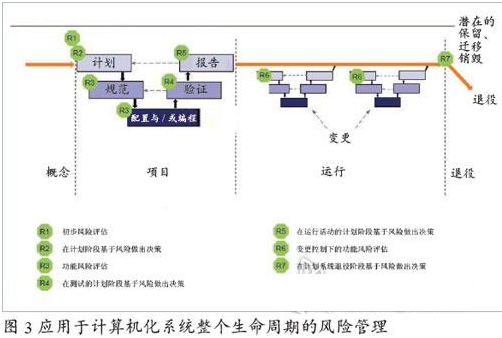
计算机化系统质量风险管理
计算机化系统的质量风险管理是一项非常重要而且有益的工作,它将贯穿于系统从初始概念提出直至最终系统退役的全过程。计算机化系统的风险管理是应用于系统整个生命周期,并针对患者安全、产品质量和数据完整性。具体如图3所示:
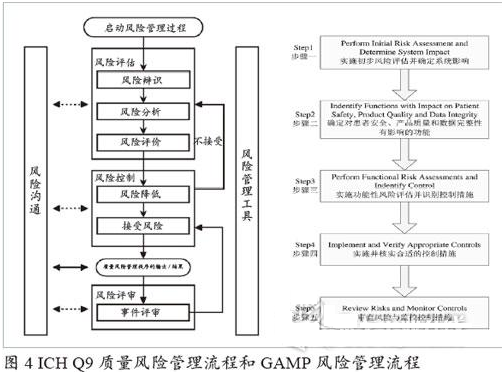
质量风险管理流程
计算机化系统质量风险管理采取了与ICH Q9质量风险管理相一致的框架进行风险评估、控制、交流与审查的系统化过程。如下图4展示了GAMP用于质量风险管理的五步流程是如何应用ICH Q9质量风险管理流程来实现和维护系统合规性。
上述的五步流程将在计算机化系统生命周期的各个阶段所实施。
风险管理工具一般采用失效模式影响分析(Failure Mode Effects Analysis, FMEA)进行计算机化系统的风险管理。
实施初步风险评估并确定系统影响:计算机化系统的初步风险评估是进行GxP关键性评估后再对GxP关键系统实施GxP影响分级。
确定对患者安全、产品质量和数据完整性有影响的功能:根据系统所要实现的功能从上述“GxP关键性”“影响级别”两个层面上进行判断和分析,从而确定并识别系统对于患者安全、产品质量和数据完整性有影响的功能。
实施并核实合适的控制措施:风险评估过程目的是采用合适的控制;依据所辨识出的风险级别可通过一组选项实现控制,这些选项包括但不局限以下:
修改工艺设计或者系统设计;
通过外部程序;
增加规范细节;
增加复查的级别和次数;
增加额外的更严格的验证活动。
风险审查与监控措施:一旦确认和实施控制,将重新进行FMEA评估,以确保风险级别得到了有效降低并且已达到可接受的水平,同时对措施情况进行持续监控。
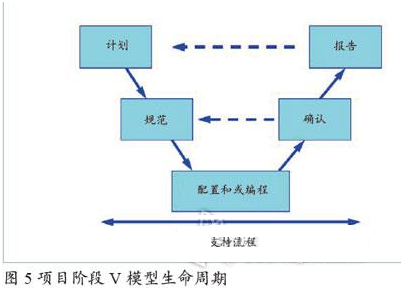
新建计算机化系统验证
基于风险的可增减的生命周期活动对系统的整个生命周期均是适用的,本节主要针对计算机化系统的验证工作在项目阶段V模型生命周期(图5)的可增减性策略。本节内容需要结合第3节和第4节一起来加以认识和理解:“软硬件分类”其实也是“质量风险管理”工作的一部分,而“可增减的生命周期活动”是“基于风险”这一基础而来的,“软硬件分类”也是其可增减的典型决策因素之一。
活动可增减性的基础和依据是质量风险管理,其决策因素主要来自如下3个方面:
系统的复杂性和新颖性(主要体现在软硬件类别和项目大小);
系统的GxP风险(对患者安全、产品质量和数据完整性的影响);
供应商评估的结果(供应商的能力水平高低)。
基于风险的可增减性策略并非是为节约成本和减少工作量寻找借口,而是以一种高效的方式合理利用资源,从而提高系统的合规效率,并更加关注于患者及公众安全这一最终目标上。
活动的可增减性主要体现在范围和深度两个层面上。对于范围而言,其可增减性主要是在项目的规范阶段和验证阶段的可伸缩;对于深度而言,整个项目的各阶段活动(包括文件及实践)深度均是一个可增减的过程。
供应商在系统的建造及合规方面扮演着非常重要的角色,因此对于供应商参与活动的平衡点也将是V模型生命周期中的一项重要工作。对于评估结果非常满意的供应商,可以考虑尽可能多的让供应商参与,从而充分利用其知识、经验和文件,用以提高系统合规效率和避免不必要的重复工作。

如下展示的各阶段的验证活动(及文件)采用的是最大的生命周期(当然,如果认为在此活动基础上风险仍不可控,则可以采取其他的或者更复杂的方式或活动来加以控制),基于系统实际的风险情况做出评估和分析后可以根据决策的结果来适当调整或减少某些活动的范围和深度。
计划阶段:编写审核并批准用户需求说明并实施初步风险评估;进行供应商评估审计并选择合适的供应商;编写审核并批准验证计划;编写审核并批准质量及项目计划。
规范阶段:编写审核并批准功能说明;编写审核并批准硬件设计说明,包括图纸;编写审核并批准软件设计说明;编写审核并批准软件模块说明;实施功能性风险评估和识别控制措施;编写审核并批准设计确认方案,执行测试及审查结果。
配置和/或编程阶段:订购硬件;构建系统;开发软件;制定配置管理计划;集成系统。
验证阶段:软件源代码审核;编写审核并批准软件模块测试方案,执行测试及审查结果;编写审核并批准工厂验收测试方案(硬件和功能);执行内部FAT预测试并审查结果(供应商内部的);执行并见证FAT测试并审查结果(被监管公司提供见证的);运至现场;安装调试;编写审核并批准SAT方案,执行测试及审查结果;编写审核并批准IQ/OQ方案,执行测试及审查结果;编写审核并批准可追溯矩阵。
报告阶段:生成系统最终文件并进行审批;保证所有设计文件均为“竣工”版本;编写技术手册;为操作人员、工程师等进行培训;生成最终验证总结报告和移交检查表并进行审查;完成移交;系统放行投入使用(运行阶段持续维护)。

遗留计算机化系统验证
近年来,由于快速发展的新技术以及监管的期望提高,同时由于GMP法规的升版等因素(如中国GMP2010版),被监管公司采取积极行动以保持其已有GxP相关系统处于验证的状态是至关重要的。
遗留系统的概念为未经验证或没有充足的证据证明其能满足现有法规要求的一个受GxP监管的运行系统。其特点主要如下:
已在生产中使用的;
不认为是满足监管期望的;
未经验证的。
验证的基本流程方法为:引入生命周期的概念;规范和验证的方法;质量风险管理。图6清晰展示了遗留计算机化系统验证的流程和方法。
小结
计算机化系统验证总体上讲就是一个“基于风险的可增减的生命周期全过程”,本文从两大方面讲述了计算机化系统验证:新建计算机化系统和遗留计算机化系统。同时对生命周期、软硬件分类、质量风险管理等一些非常重要的知识、方法或理念进行了较为详尽的介绍。
参考文献
[1] 国家食品药品监督管理局.药品生产质量管理规范(2010年修订).2010.
[2] US Code of Federal Regulations, Title 21, Food
and Drugs, 21CFR Part 11,Electronic Records, Electronic Signatures, 2011.
[3] ISPE GAMP5:A Risk-Based Apporach to Compliant
GxP Computerized Systems,International Society for Pharmaceutical
Engineering,Fifth Edition,February 2008. www.ispe.org
[4] EudraLex Volume 4 EU Guidelines to Good
Manufacturing Practice – Medicinal Products for Human and Veterinary Use,Annex
11- Computerized Systems, June 2011.
[5] ICH Q9 Quality Risk Management, November 2005,
4th phase.
如有计算机化系统管理方面的问题,可向长沙金领医药科技有限公司尹显峰咨询,手机号:15973123761。
上一篇:无
下一篇:计算机化系统软件分类与验证原则!